Marketing a medical device in the EU requires the preparation of technical documents that demonstrate compliance with the Medical Device Directive (MDD), the Medical Device Regulation (MDR), and other requirements as well as the implementation of specified conformity assessments.
SunFlare provides prompt and reliable consulting services related to the acquisition of CE marking. These services range from identifying the basic requirements that are applicable to the client through to drafting technical files (Clinical Evaluation Reports, etc.).
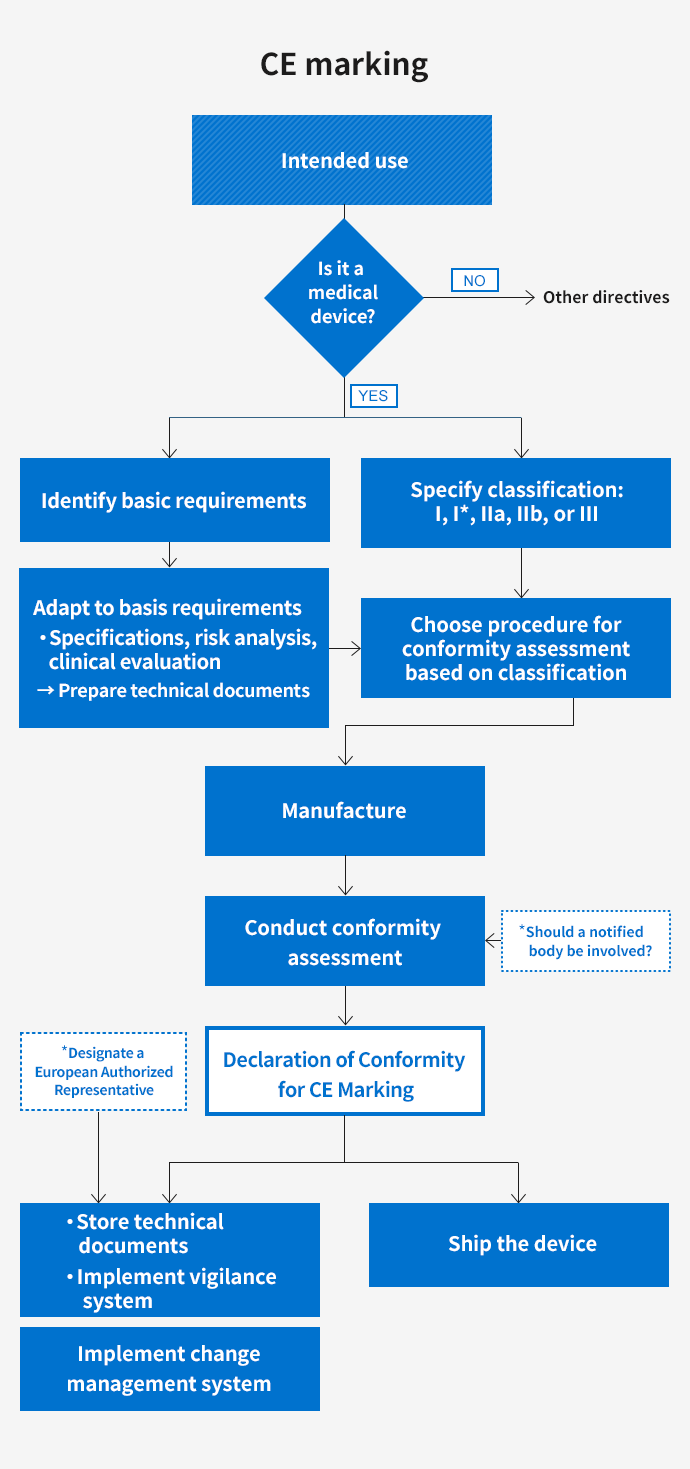
Workflow for complying with the MDD and other regulations for European CE marking
Projects conducted over the past 11 years to support the acquisition of CE marking
Consulting services provided by SunFlare
CE marking requirements
We provide consulting services related to identifying differences between Japanese and European regulations to enable our clients to obtain CE marking. As part of these services, we:
Investigate the requirements (identify design inputs, including harmonized standards)
Analyze the client’s product to determine whether it complies with the requirements for CE marking
Hold lectures on the requirements
Compliance with CE marking requirements
Design development (essential requirements, product classification, applicable standards, etc.)
Clinical evaluations
Post-marketing surveillance (PMS)
Software lifecycle processes (EN 62304)
Usability engineering (EN 62366)
Risk management (EN 14971)
Labeling (EN ISO 15223-1, EN 1041 etc.)
Vigilance system
Sterilization validation and sterile barrier system (related to transport simulation testing in accordance with ISO 4180, etc.)
Provide and customize samples of the following procedure manuals
Procedure manuals related to CE marking
Documents related to clinical evaluations:
Clinical evaluation plans, clinical evaluation reports, literature search protocols, literature search reports, etc.
Assessment reports on non-compliance with EU risk management requirements
Procedure manuals for preparing technical documents
Samples of Part A of technical files and compliance statements (templates)
Vigilance procedures and forms that comply with European standards
Usability engineering procedure manuals and forms:
Specifications, verification plans, verification reports, validation plans, and validation reports
Software lifecycle process procedure manuals and forms:
Safety classification reports, development plans, requirement definitions, architecture specifications, testing protocols, and testing reports
Transition to the Medical Devices Regulation (MDR)
We provide consulting services related to transitioning from the EU’s Medical Device Directive (MDD) to its Medical Devices Regulation (MDR). As part of these services, we:
Hold on-site seminars related to MDR requirements
Analyze the client’s product to determine whether it complies with CE marking requirements (provision of information about differences between the MDD and the MDR)
Work with the client to examine action plan drafts and related documents, including documentation related to the following:
General Safety and Performance Requirements (GSPR)
Procedural steps for ensuring compliance with CE marking
Quality manuals
Technical files
Post-marketing surveillance (PMS) reports and periodic safety update reports (PSUR)
Labeling
Contracts and agreements
Provide related sample documents and review documents prepared by the client
We provide a range of services related to the conducting of clinical evaluations, from devising clinical evaluation strategies and conducting academic paper research to drafting clinical evaluation reports. In providing these services, we carry out the following steps:
Creating a new clinical evaluation plan (in compliance with MEDDEV 2.7/1 Rev.4)
Step 1: Analyze the product’s future prospects and assess the evaluation methods
Hold lectures on the requirements for MEDDEV 2.7/1 Rev. 4
Check the current risk management, non-clinical evaluations, and related documentation
Investigate equivalent products and verify their equivalence
Conduct a preliminary literature investigation to identify core clinical data
Draft a plan, if feasible, based on the above activities
Step 2: Draft documents related to the clinical evaluation
Have the clinical evaluation report translated by specialists
Provide advice on responding to inquiries from the notified body
Updating from Rev. 3
Hold lectures on the requirements for MEDDEV 2.7/1 Rev. 4
Check the current clinical evaluation report and related documents
Provide the following sample documents (in compliance with MEDDEV 2.7/1 Rev. 4):
Clinical evaluation report, declaration of interests form, assessment form for determining the necessity of a post-market clinical follow-up (PMCF), and clinical evaluation planning form
Assist in adapting the content of the client’s existing clinical evaluation report to the format required under MEDDEV 2.7/1 Rev. 4, summarize missing items, etc.
Provide feedback and advice on items that need to be corrected or added
Provide advice on responding to inquiries from the notified body
Over the past 9 years, we have carried out 190 projects related to supporting the preparation of clinical evaluation reports that comply with MEDDEV 2.7/1 Rev. 4 and MDCG guidances.
Main target products Diagnostic imaging devices, rehabilitation devices, nursing-care devices, hemodialysis units, catheters, stent grafts, dental materials, dental devices, hemostats, synthetic bone, suture thread and other new medical devices
We assist in establishing a quality management system (QMS) that complies with the European Standard EN ISO 13485. As part of this assistance, we:
Analyze the client’s existing QMS to determine whether it complies with the requirements of EN ISO 13485
Provide and customize samples of procedure manuals and forms related to acquiring CE marking, conducting clinical evaluations, preparing technical documents, and ensuring vigilance compliance with European standards
In some ASEAN countries, obtaining CE marking for a product can simplify the examination process and provide a partial exemption for the document submission requirements. Therefore, obtaining CE marking facilitates access not only to the EU but also to the ASEAN market.
Drafted by the European Commission in 2012, the Medical Device Regulation (MDR) was adopted on April 5, 2017, to replace the Medical Device Directive (MDD) as the EU’s regulatory framework for medical devices. This regulation was officially enacted on May 26, 2017. For details,please see the MEDTEC WEB website (in Japanese). SunFlare’s monthly e-mail magazine on medical devices also provides information on the latest trends related to the MDR, other laws and regulations in various countries, and revisions to standards. This e-mail magazine is available only in Japanese. To subscribe, please fill out the registration form.