サン・フレアのコンサルティング内容
CEマーキングの要求事項に関するコンサルティング
CEマーキングに伴う日本とヨーロッパにおける具体的なギャップ対応に関するコンサルティングを行います。
要求事項の調査(整合規格等の設計インプットや関連するガイドライン等の明確化)
要求事項と貴社ご状況とのギャップ分析およびギャップ対応に関する助言提供
要求事項のレクチャー
CEマーキング対応全般
設計開発プロセスにおける要求事項(GSPR(*)、クラス分類、適⽤規格等)
臨床評価/性能評価
ソフトウェアライフサイクルプロセス(EN 62304)
サイバーセキュリティ(EN IEC 81001-5-1)
ユーザビリティエンジニアリング(EN 62366-1)
リスクマネジメント(EN ISO 14971)
ラベリング(EN ISO 15223-1, EN 1041 etc.)
市販後調査(PMS:Post-Market Surveillance)
ビジランスシステム
安定性試験
生物学的安全性評価(EN ISO 10993シリーズ)
滅菌バリデーションおよび無菌バリアシステム(EN ISO 11607-1, -3/輸送シミュレーション試験(ISO 4180等への準拠)関連等)
製造業者、指定代理人(法定代理人)、ノーティファイドボディ、輸入業者、販売業者等の事業者の責務
下記手順書サンプルの提供・カスタマイズ
CEマーキングに関する手順書
臨床評価/性能評価関連文書:
欧州リスクマネジメントギャップ検討書
技術文書作成手順書
MDR/IVDRの要求事項に基づいたテクニカルドキュメンテーション(技術文書)(STED形式)
ユーザビリティエンジニアリング手順書および様式:
ソフトウェアライフサイクルプロセス手順書および様式:
欧州向けビジランス手順書および様式
市販後調査(PMS)における手順・様式および計画書
市販後臨床フォローアップ(PMCF)/市販後性能フォローアップ(PMPF)
目次に戻る
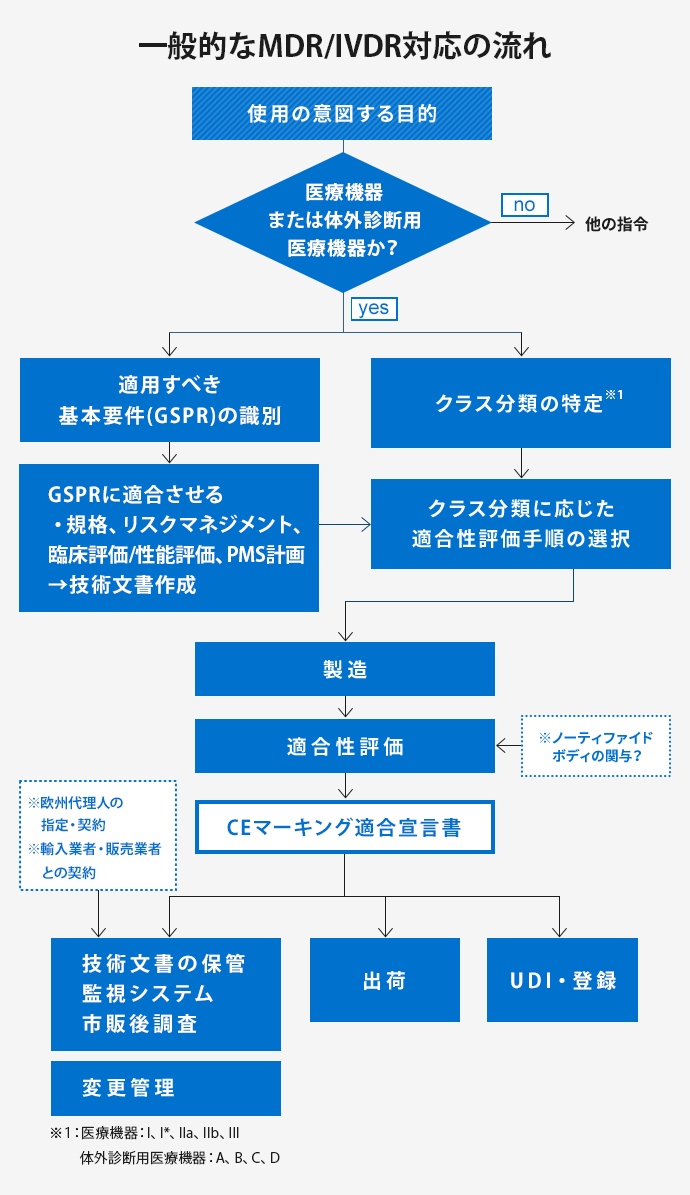
欧州医療機器規則(MDR)/欧州体外診断用医療機器規則(IVDR)
2017年4月5日、ヨーロッパにおける医療機器の規制枠組みであった医療機器指令(MDD)、能動型植込み可能医療機器指令(AIMDD)に代わり、2012年に欧州委員会で起案された医療機器規則(MDR)が可決され、新型コロナウイルスの感染拡大の影響よる1年延期ののち、2021年5月26日より施行されています。また、体外診断用医療機器については、体外診断用医療機器指令(IVDD)に代わり、体外診断用医療機器規則(IVDR)が、2022年5月26日より施行されています。
MDRおよびIVDRでは、これまでの指令に比べ、マニュファクチャラーをはじめとするEconomic operatorやノーティファイドボディ等のステークホルダーに関する要求事項や、製品自体に係る要求事項が厳格化されており、対応にあたっては、MDR/IVDR本体はもちろん、関連する主要なガイダンス等について内容をきちんと把握・理解することが非常に重要です。
サン・フレアでは、欧州CEマーキング支援およびMDR/IVDR対応に関するコンサルティングサービスの他、以下のような情報提供を本Webサイトにて行っております。皆様の日常業務に、どうぞご活用ください。
医療機器指令/MDD:Medical Device Directive
■規制対応のための重要資料(MDR/IVDR)
■サン・フレア 医療機器メールマガジン 登録フォーム よりご登録ください。
目次に戻る
欧州医療機器規則(MDR)/体外診断用医療機器規則(IVDR)移行支援
旧指令(MDD/AIMDD/IVDD)から医療機器規則(MDR)/体外診断用医療機器規則(IVDR)への移行支援を行います。
MDR/IVDR要求事項に関するオンサイト/オンラインセミナー
要求事項と貴社ご状況とのギャップ分析(旧指令との差分情報の提供)
対応計画案および関連する文書の共同検討(例えば以下のような項目について)
GSPR
CEマーキング対応手順
品質マニュアル
テクニカルドキュメンテーション(技術文書)
市販後調査計画に関する文書(PMS、PSUR: Periodic Safety Update Report)
ラベリング
契約類
上記関連サンプル文書提供および貴社作成文書のレビュー
目次に戻る
臨床評価/性能評価の実施に関するコンサルティング
臨床評価/性能評価の戦略の策定、文献抽出から臨床評価報告書/性能評価報告書のドラフティングまで行います。
目次に戻る
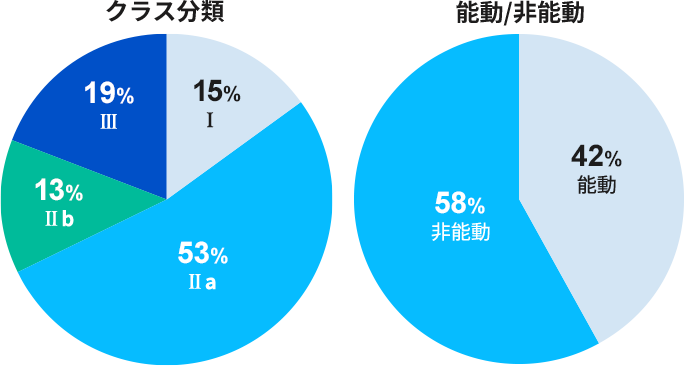
対応実績(臨床評価)
過去8年間で179件 のMEDDEV 2.7/1 rev.4 および関連MDCG文書準拠の臨床評価報告書作成支援実績があります。
主な対象製品
画像診断関連機器、リハビリ/介護用機器、透析装置、カテーテル(中心循環系用を含む)、ステントグラフト、ガイドワイヤー、眼科用機器、歯科材料、歯科用機器、止血剤、人工骨、縫合糸、脳神経外科用機器、心臓血管外科用機器、医療機器ソフトウェア、その他新医療機器 etc…
目次に戻る
テクニカルドキュメンテーション(技術文書)のドラフティングに関するコンサルティング
テクニカルドキュメンテーション(技術文書)のドラフティングを行います。
テクニカルドキュメンテーション(技術文書)のドラフティング
MDR/IVDRの要求事項に基づいたテクニカルドキュメンテーション(STED形式)のドラフティング
テクニカルドキュメンテーションの高い専門性を備えた翻訳
ノーティファイドボディによる審査への対応
目次に戻る
欧州要求事項に適合した品質マネジメントシステム(EN ISO 13485+欧州追加要求事項)の構築・運用に関するコンサルティング
ヨーロッパ対応のQMS構築支援を行います。
現行品質マネジメントシステム構築状況の確認、欧州追加要求事項とのギャップ分析
CEマーキング、臨床評価/性能評価、技術文書作成、欧州向けビジランスの手順書および様式の文書サンプル提供およびカスタマイズ
目次に戻る
指定代理人(法定代理人)の選定支援に関するコンサルティング
独自の調査資料に基づく指定代理人(法定代理人)の選定支援を行います。
指定代理人(法定代理人)候補の選定支援およびリストのご提供
選定に関する助言提供
目次に戻る
市販後安全管理活動に関するコンサルティング
ヨーロッパにおける市販後安全管理活動支援を行います。
欧州ビジランスシステムの構築
対象製品および類似製品等の各国不具合情報収集
MDR/IVDRに準拠した市販後調査(PMS)手順の作成支援1) ギャップ分析
現行PMS手順および関連資料の確認
現行手順とMDR/IVDR要求事項のギャップ分析および手順案作成方針の検討と検討結果の報告
2) 手順・様式の改訂案のドラフト
1)の検討に基づく貴社PMS手順・様式の改訂案の提供と説明
必要に応じた手順・様式改訂案の修正
※サン・フレアでは、日本における市販後対応(GVP、不具合報告等)に関するコンサルティングも承っております。サービス内容の詳細 はこちらのページをご確認ください。
目次に戻る
ASEAN 諸国とCEマーキングの関係性について
ASEAN諸国では、製品をCEマーキングしていることで、審査の簡略化、提出資料の一部免除等の方針を採用している国があります。CEマークの取得により、ヨーロッパはもちろんのこと、ASEAN市場へのアクセスがより身近なものとなります。
※サン・フレアでは、日本における承認申請時の臨床評価に関するコンサルティングも承っております。サービス内容の詳細はこちらのページ をご確認ください。
目次に戻る